Projects
Proteomics
Mass spectrometry
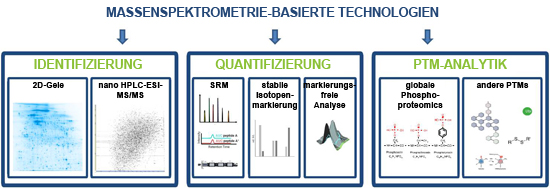
The key technology involved in identifying and quantifying proteins in biological samples is the biological mass spectrometry. The field of application of this technology exceeds the mere possibility of identifying proteins in 2D-gel spots. By means of a so called mark-free analysis, it is possible to globally identify and quantify proteins from very small samples, without the use of 2D-gels. This happens due to its intensities in the LC-MS run, in which different samples are compared to each other directly. Another option for the exact quantification on a global level is the use of stable, heavy isotopes. This way two or more samples can be mixed and compared to one another in one and the same run, since the masses of identical proteins differ in different samples, according to the applied isotope-marking. A possibility for the targeted, very exact and absolute quantification of single (candidate-) proteins is the SRM-AQUA method, in which chosen synthetic isotope-marked peptides (analogues of native peptides of the protein) are mixed into the sample as the internal standard in a defined quantity, and then specifically detected in the mass spectrometer. In addition to this, phosphorylation, which plays an essential role in the regulation and signal transduction of many cellular processes and can be relevant to a disease, can be analyzed and quantified globally. Other posttranslational modifications, such as for example acetylation or glycosylation can be detected with mass spectrometrical methods as well.