Projekte
Biospektroskopie
Bioinformatik
Die computergestützte Analyse spielt eine zentrale Rolle bei der Aufdeckung von Biomarkern – bei der bildgebenden Spektroskopie in Geweben ebenso, wie bei der Spektroskopie von Körperflüssigkeiten. Um in den komplex strukturierten spektralen Datenmengen krankheitsspezifische Merkmale zu identifizieren, kommen algorithmische Methoden der Mustererkennung zum Einsatz, die spektrale oder morphologische Merkmale zur Unterscheidung zwischen gesunden und krankheitsbetroffenen Proben ermitteln. Dabei kommen auch sehr aktuelle Methoden des sogenannten Deep Learnings zum Einsatz. Ein genaueres Gesamtbild eines Krankheitsmusters ergibt sich oft, wenn Bild- oder Spektraldaten aus unterschiedlichen Mikroskopen oder Messmethoden kombiniert werden. Solche Cross-Plattform Analysen stellen wichtige Herausforderungen an die Bioinformatik dar und bilden dementsprechend einen Schwerpunkt der Bioinformatik in PURE.
Um die anfallenden großen Datenmengen mit den rechenintensiven Methoden der Mustererkennung zu verbinden ist zudem eine IT-Infrastruktur aus Dateiservern und Cluster-Computern notwendig, die von Seiten der Bioinformatik betrieben wird.
Markerfreie Identifikation von Zelltypen und subzellulären Strukturen mit Hilfe der Ko-Lokalisations-Analyse
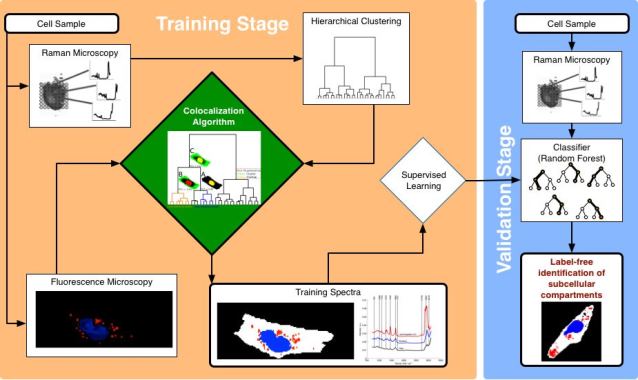
Abb. 1: Schematischer Ablauf der Ko-Lokalisiationsanalyse zum markerfreien entschlüsseln subzellulärer Strukturen.
Ein entscheidender Vorteil der Raman-Mikroskopie ist die Möglichkeit, Proben markerfrei untersuchen zu können. Die Ortsauflösung ist dabei so hoch, dass sich eine einzelne Zelle mit zehntausenden von Pixeln darstellen lässt. Die Raman-Spektren an jeder Pixelposition sind sehr charakteristisch für die einzelnen Bestandteile einer Zelle, so dass die subzellulären Strukturen in Raman-Bilddaten aufgeschlüsselt werden können.
Die spektralen Eigenschaften einzelner subzellulärer Bausteine müssen jedoch zunächst statistisch gelernt werden. Hierzu werden Raman-Bilder mit Fluoreszenz-Bildern der gleichen Zelle überlagert. An genau dieser Stelle setzt die in PURE entwickelte Ko-Lokalisations-Analyse an. Diese extrahiert repräsentative Spektren für Zellkomponenten, in denen das Fluoreszenzbild und das Raman-mikroskopische Bild korrelierte Strukturen aufweisen.
Das Ergebnis der Ko-Lokalisations-Analyse ist ein Klassifizierer, der die subzelluläre Struktur einer Zelle schließlich vollständig markerfrei -- nur anhand des Raman-mikroskopischen Bildes -- entschlüsseln kann. Insbesondere eignen sich solche Klassifizierer auch, um Zelltypen zu unterscheiden, und können daher in der Zytopathologie verwendet werden, um kranke von gesunden Zellen, die etwa im Urin zu finden sind, unterscheiden zu können.
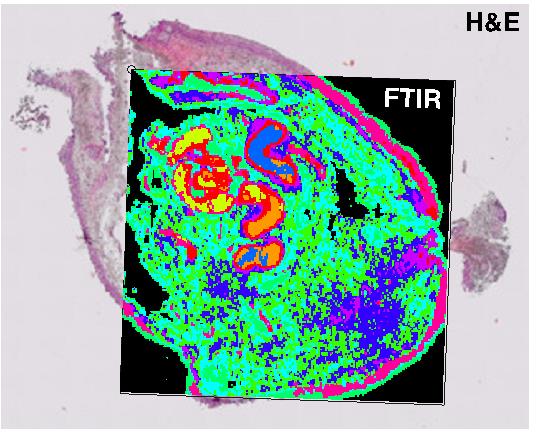
Abb. 2: Ergebnis einer Bild-Registrierung. Die Koordinaten des Infrarot-mikroskopischen Bildes (FTIR) wurden mit denen des histopathologischen Färbungsbildes (H&E) exakt und vollautmatisiert überlagert.
In PURE wie auch in anderen Studien ist es übliche Praxis, dass eine Probe mit verschiedenen Arten von Mikroskopen untersucht wird. Oft wird beispielsweise eine Probe zunächst infrarotmikroskopisch vermessen, und anschließend nach einer histopathologischen Färbung mit einem konventionellen Lichtmikroskop erfasst. Um die entstehenden sehr verschiedenartigen Bilddaten analysieren zu können, ist ein entscheidender Vorverarbeitungsschritt notwendig: jede Koordinate im Infrarot-Bild sollte derselben Position in der Probe entsprechen, wie die identische Koordinate im lichtmikroskopischen Bild. Der entsprechende Koordinaten-Transfer kann automatisiert von einem sogenannten Registrierungs-Algorithmus durchgeführt werden.
Dieser Algorithmus muss Merkmale aus sehr unterschiedlich gearteten Bilddaten in Einklang bringen, und gleichzeitig mit Bilddaten zurechtkommen, die bis zu 100 Gbyte in einem Datensatz umfassen können. In PURE wurde in der AG Bioinformatik ein entsprechender, sehr robuster und gleichzeitig effizienter Registrierungs-Algorithmus entwickelt.
Storage Infrastruktur mit Gesamtkapazität von >400 TByte
High-Performance-Computing:
> Rechenkapazität mit insgesamt >1500 CPU-Kernen
> HPC-Rechner mit bis zu 1 TByte Hauptspeicher
> GPU-Rechner